Dr Jorge Melendez Zajgla, Dra Vilma Maldonado
La ingeniería genética, un tema que llama mucho la atención, se ha convertido en un tema polémico en los últimos años. La edición genética, que es una de las áreas más interesantes y éticamente complicadas, tiene el potencial de curar enfermedades que tengan un componente genético, pero también levanta preocupaciones sobre tener “niños a medida”, además de la posibilidad de generar consecuencias imprevistas. En este artículo voy a repasar lo más importante sobre la edición genética.

Created with AI using Stable Diffusion and DALL-E 2. OpenArt
¿Qué es la Edición Genética?
La edición genética es una técnica utilizada para modificar el ADN de un organismo. Implica la inserción, eliminación o reemplazo de secuencias de ADN. El método más común de edición genética es el sistema CRISPR-Cas9. Otros métodos incluyen nucleasas de dedos de zinc y TALENs.
Beneficios potenciales de la edición genética
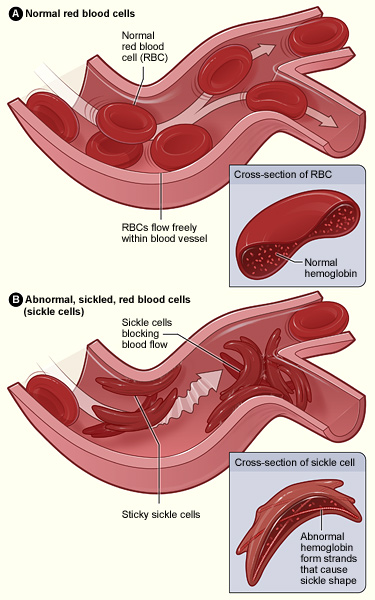
Uno de los aspectos más emocionantes de la edición genética es su potencial para curar enfermedades genéticas. La edición genética podría usarse para eliminar o reparar los genes mutados responsables de enfermedades como la fibrosis quística y la anemia falciforme. La edición genética también podría usarse para crear cultivos y ganado más resistentes a enfermedades o al cambio climático. ¡En un futuro distante podría incluso ser la clave para la supervivencia humana, ya sea dentro o fuera de la tierra!
Ejemplos del mundo real de los beneficios potenciales de la edición genética incluyen el uso de CRISPR para curar a pacientes con enfermedad de células falciformes y el desarrollo de mosquitos modificados genéticamente que son resistentes a la malaria.
La edición genética se puede dividir en dos grupos: germinal y somática. En el caso de la primera, el cambio en el ADN se realiza en los óvulos o en embriones tempranos, lo que hace que la edición no solo se presente en todas las células del organismo, sino que este cambio se hereda a las futuras generaciones. En el caso de la edición somática, el cambio se realiza a un grupo de células únicamente, lo que hace que no sea heredado.
Consideraciones éticas
El uso de la edición genética en la línea germinal humana plantea una serie de preocupaciones éticas. Una de las preocupaciones más significativas es la de los “bebés diseñados”. La edición genética podría usarse para seleccionar rasgos como la inteligencia o la apariencia física, lo que plantea preguntas sobre la ética de manipular el material genético de las generaciones futuras. Otra preocupación es el potencial de consecuencias no deseadas. La edición genética podría tener efectos imprevistos en el medio ambiente o en la salud del organismo que está siendo editado.
En el caso de la edición genética somática en el ser humano, las preocupaciones se limitan a la posibilidad de efectos secundarios, dado a que potencialmente se podrían presentar cambios en lugares del ADN diferentes.
Finalmente, la edición genética de organismos no humanos tiene consideraciones éticas basadas en los posibles efectos sobre el medio ambiente y la ecología específica del organismo modificado.
Tipos de técnicas de edición genética
Existen diferentes técnicas de edición genética, cada una con sus propias fortalezas y debilidades. El método más común es el sistema CRISPR-Cas9, que utiliza una enzima bacteriana para cortar el ADN en una ubicación específica. Otros métodos incluyen nucleasas de dedos de zinc y TALENs.
Es importante considerar cuidadosamente los diferentes tipos de técnicas de edición genética y sus posibles aplicaciones. Por ejemplo, CRISPR tiene un alto grado de precisión y especificidad, pero también puede tener efectos fuera del objetivo. Existen múltiples grupos de investigación que se encuentran mejorando las técnicas para evitar los efectos fuera de la región del ADN blanco.
Estado actual de la investigación de edición genética
Aunque la edición genética ha sido marcada por preocupaciones éticas en el pasado, tiene el potencial de revolucionar la medicina y mejorar la vida de millones de personas. La edición genética es un campo que avanza rápidamente, y hay una serie de proyectos de investigación en curso que exploran sus posibles aplicaciones. Algunas de las áreas más prometedoras de investigación incluyen el uso de la edición genética para curar enfermedades genéticas hereditarias y no hereditarias, como el cáncer. Actualmente hay más de 50 estudios experimentales de edición genética en marcha para tratar desde el cáncer hasta el VIH y enfermedades de la sangre.
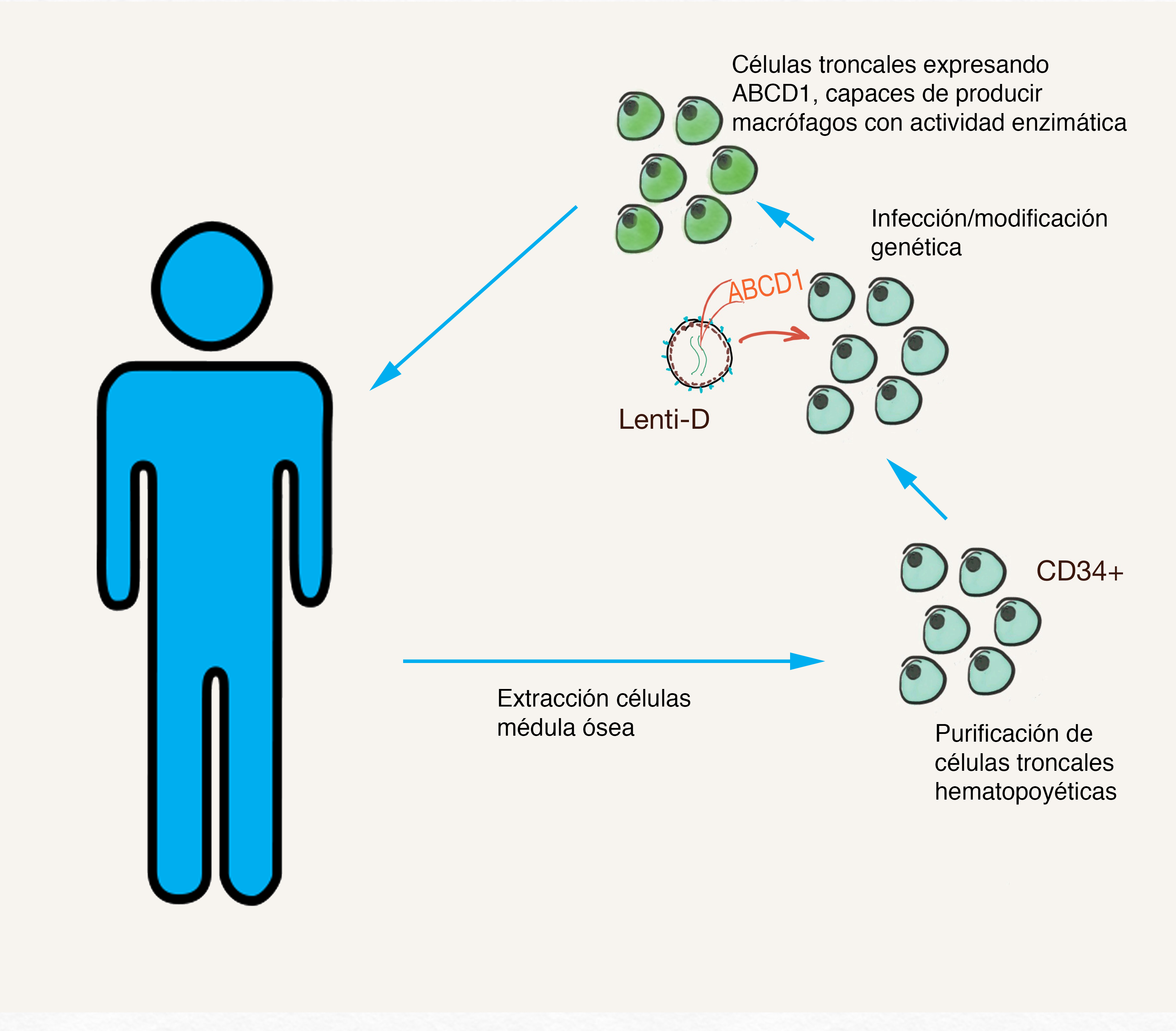
Un ejemplo de esto es el reciente desarrollo de un tratamiento para la anemia de células falciformes por la compañía Vertex basado en la modificación genética por CRISPR. En esta técnica, los afectados reciben un duro tratamiento de quimioterapia para remover la médula ósea para realizar un transplante. Células extraídas antes de la quimioterapia se modifican para corregir el defecto en el ADN causante de la anemia antes de ser transplantadas en los pacientes. La compañía ha tratado a 75 pacientes de esta manera, y nos encontramos esperando ansiosamente la publicación de los resultados finales. Sin embargo, ya existen reportes anecdóticos, como el de Victoria Gray, que platicó su experiencia en la Tercera Reunión Internacional de la Edición Genética Humana. Esta paciente sufría episodios importantes de dolor que la mantenían hospitalizada por meses. El transplante de lo que ella denomina “supercélulas” le permitió cambiar esto, llevándola a comentar que “Me encuentro aquí frente a ustedes como una prueba de que los milagros existen”(1).
Existe un gran potencial para el uso en muchas otras enfermedades, como es el caso del cáncer, en el que se esperan resultados próximos. Incluso, la FDA de Estados Unidos le ha otorgado la designación de Terapia Avanzada de Medicina Regenerativa (RMAT por sus siglas en inglés) al medicamento CTX130, de la compañía CRISPR Therapeutics, que consiste en un una terapia con células inmunes modificadas por CRISPR. Estas células se han modificado para dirigirlas contra un tipo específico de tumor, denominado linfoma cutáneo de células T (2).
Created with AI using Stable Diffusion and DALL-E 2. OpenArt
Paisaje regulatorio
El paisaje regulatorio que rodea a la edición genética germinal es complejo. Actualmente, el uso de la edición genética está regulado de manera diferente en diferentes países. En los Estados Unidos, la FDA regula los productos de terapia génica, pero no hay una regulación específica de la edición genética. En México no existe avance al respecto, hasta el momento. En el caso de la regulación para la terapia somática, esta cae dentro del marco actual, por lo que no representa mayores problemas que los que se tiene para terapia celular actual.
Es importante comprender el paisaje regulatorio que rodea a la edición genética para navegar las consideraciones legales y éticas de su uso.
Impacto potencial en la sociedad y la economía
El impacto potencial de la edición genética en la sociedad y la economía es significativo. La edición genética podría revolucionar la atención médica al proporcionar curas para enfermedades actualmente incurables o incluso eliminar enfermedades transmitidas por vectores. También podría crear cultivos y ganado más resistentes, lo que conduciría a un aumento de la seguridad alimentaria, ayudándonos a enfrentar el cambio climático. Sin embargo, la edición genética conlleva una serie de riesgos potenciales que necesitan ser estudiados, como se mencionó antes y que dependen del fin específico que se busca. No es lo mismo realizar edición en la línea germinal que en la somática, ni la edición de un cultivo domesticado que en un vector que transmite la malaria. Cada una de estas aplicaciones debe de ser estudiada profundamente. Finalmente cabe mencionar que el costo de la aplicación de esta herramienta puede hacerla inaccesible a la mayoría de la población. El desarrollo de esta tecnología de manera local podría ayudar a disminuir costos, haciéndola accesible a nuestra población. En el futuro mediato se estará utilizando en los países con economías avanzadas, por lo que sería mejor no tener que importarlas a alto costo si es posible desarrollarlas en nuestro país.
Bibliografía








")